Computational Biophysics in Drug Discovery
Dr. Janmejaya Rout
February 15, 2026
Introduction
Computational biophysics lies at the intersection of physics, mathematics, and computer science, offering powerful tools to probe biological systems at atomic resolution. In drug discovery, it has become indispensable, enabling researchers to simulate and predict how candidate molecules interact with complex biological targets such as proteins, nucleic acids, and membranes.
Traditional drug development often relies on large-scale experimental screening, a process that is both costly and time-intensive. In contrast, computational approaches, particularly molecular docking and molecular dynamics (MD), have transformed the field. MD simulations provide a dynamic view of biomolecules, bridging the gap between static crystal structures and the fluid reality of living systems. When combined with docking, enhanced sampling, and free energy calculations, these methods allow scientists to explore conformational changes, binding mechanisms, and thermodynamic stability before advancing compounds to laboratory testing.
Molecular Docking
Molecular docking is a cornerstone of Structure-Based Drug Design (SBDD), providing a computational framework to predict how small molecules orient and interact with biological targets. By estimating binding poses and affinities, docking accelerates the identification and refinement of drug candidates before costly experimental validation. The availability of high-resolution structural data from X-ray crystallography or cryo-electron microscopy in repositories like the Protein Data Bank has further strengthened its utility. Molecular Docking is most commonly used for target identification, virtual screening, lead optimization, and drug repurposing.
- Target identification: Docking helps researchers locate active or allosteric sites on proteins.
- Virtual screening: Millions of compounds can be computationally tested, narrowing down candidates before experimental validation.
- Lead optimisation: Chemists can tweak molecules to improve potency, selectivity, and reduce side effects.
- Drug repurposing: Approved drugs can be screened against new targets, a strategy that proved vital during recent health crises such as Covid-19.
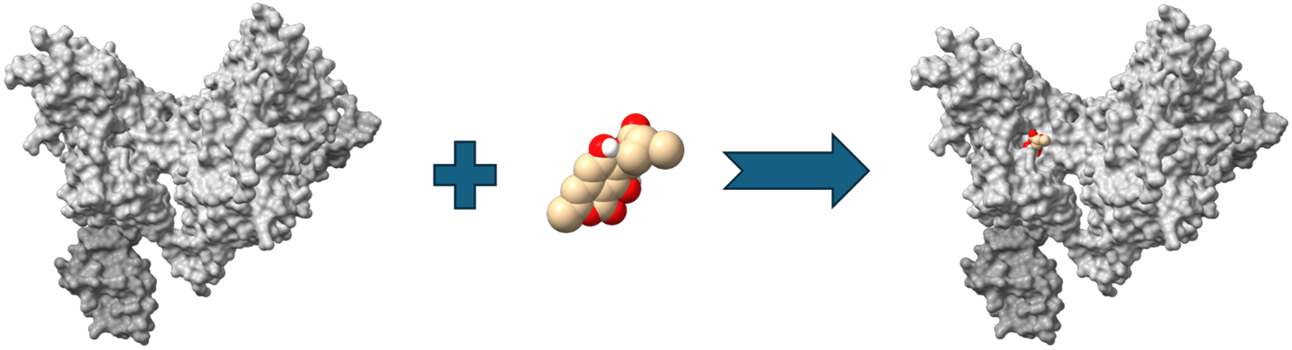
Molecular Dynamics
Proteins are not rigid structures. They bend, twist, and shift into different shapes. Molecular Dynamics (MD) simulations capture this motion by solving Newton’s equations of motion, offering a time-resolved and atomistic view of biomolecular systems. This enables exploration of protein flexibility, ligand-binding dynamics, conformational transitions, and thermodynamic properties.
- Protein flexibility: MD reveals hidden binding pockets and allosteric mechanisms.
- Docking refinement: It tests whether predicted binding poses remain stable in realistic, solvent-filled environments.
- Binding free energy calculations: Methods like MM-PBSA and Free Energy Perturbation (FEP) provide quantitative estimates of ligand affinity, often with near-experimental accuracy.
- Enhanced sampling: Techniques such as metadynamics or replica exchange MD help explore rare events like ligand unbinding or protein folding/unfolding.
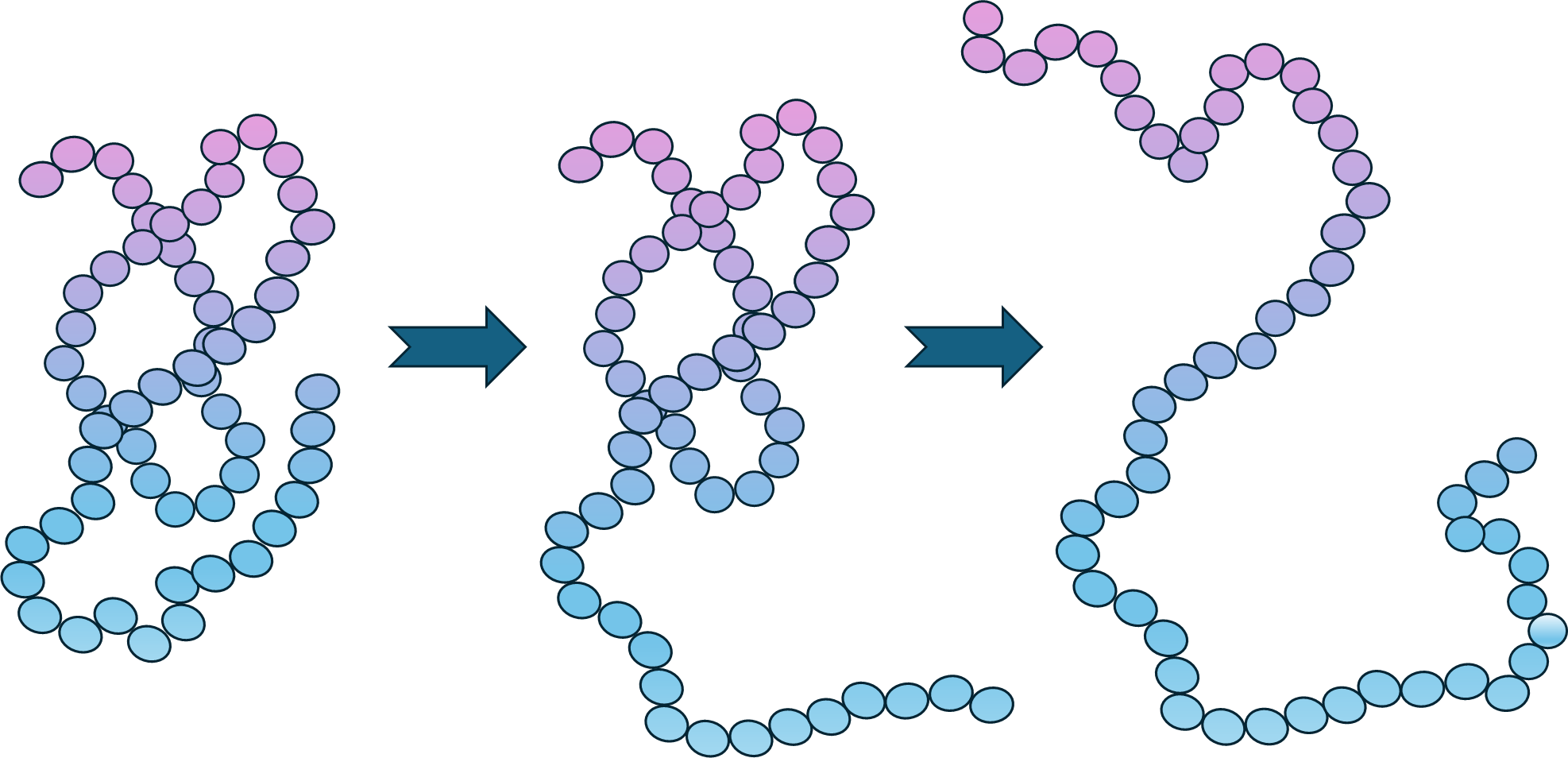
Applications
Docking and MD are now integrated into nearly every stage of drug development including
- Hit validation: Confirming stability of docked complexes.
- Lead optimization: Predicting affinity changes with FEP.
- Selectivity profiling: Comparing ligand binding across related proteins.
- Mechanistic insights: Understanding resistance mutations.
- Membrane protein modeling: Simulating GPCRs and ion channels in lipid bilayers.
Pharmaceutical companies increasingly rely on MD studies, especially FEP workflows as decision-making tools that reduce experimental cycles and accelerate development timelines.
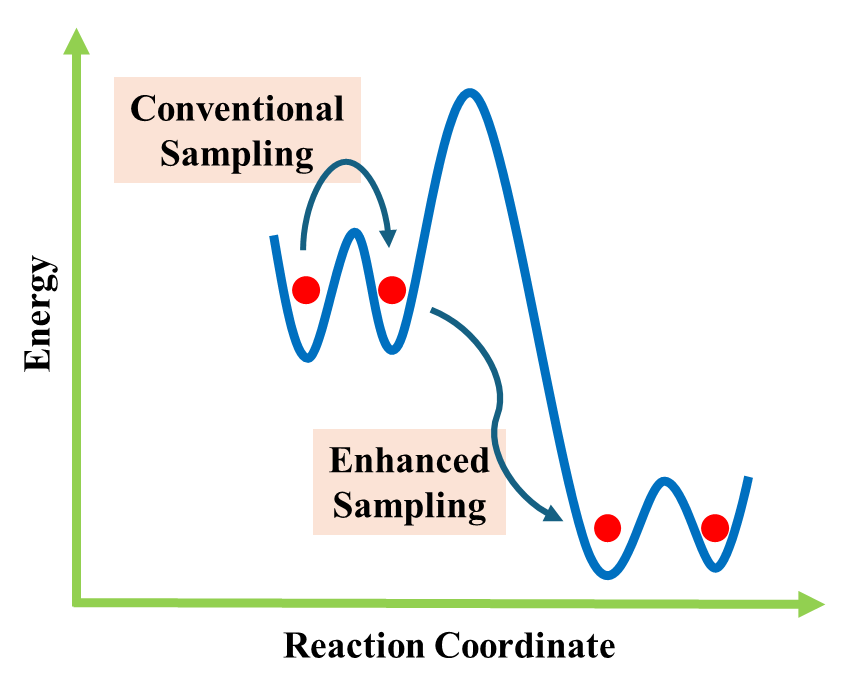
Challenges and Future Directions
Despite its transformative role, computational biophysics faces challenges.
- Docking can oversimplify protein flexibility and solvent effects.
- MD requires significant computational resources.
- Force field accuracy and sampling limitations remain hurdles.
Though there are challenges, advances in GPU computing, improved force fields, and hybrid physics-based machine learning approaches are steadily overcoming these barriers. The integration of AI with biophysics promises even greater accuracy and scalability in the near future.
Conclusion
Computational biophysics is no longer just a supporting tool. It has become a driving force in modern drug discovery. Docking provides speed and efficiency, while MD adds realism and depth. Together, they enable scientists to design drugs more intelligently, reducing experimental costs to tackle targets once thought “undruggable.” With ongoing advances in high-performance computing and machine learning, computational biophysics will continue to play a pivotal role in guiding pharmaceutical innovations.
References
- Kitchen, D., Decornez, H., Furr, J. et al. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3, 935–949 (2004).
- Hollingsworth, Scott A. et al. Molecular Dynamics Simulation for All.Neuron, 99(6), 1129 - 1143 (2018)
- Wang, L., et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. JACS, 137(7), 2695–2703 (2015).
- Lionta, E., et al. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Current Topics in Medicinal Chemistry, 14(16), 1923–1938 (2014).
- Sliwoski, Gregory et al. Computational Methods in Drug Discovery. Pharmacological Reviews, 66(1), 334–395 (2014).